Artículo de revisión
Mecanismos moleculares del síndrome de Down
Molecular mechanisms of Down syndrome
Zury Sadai Valdivieso Solorzano 1 * https://orcid.org/0009-0006-7328-8262
Dayana Marisol Aldas Acosta 1 https://orcid.org/0009-0002-0353-4801
Daniel Esteban Padilla Barragán 1 https://orcid.org/0009-0006-6622-8325
1 Universidad Regional Autónoma de Los Andes, Ambato. Ecuador.
*Autor para la correspondencia. Correo electrónico: zuryvs64@uniandes.edu.ec.
RESUMEN
El síndrome de Down es una condición genética caracterizada por la presencia de una copia extra del cromosoma 21. La embriología del síndrome de Down ha sido objeto de intensas investigaciones para comprender cómo esta alteración cromosómica afecta el desarrollo embrionario y fetal. A lo largo de las primeras etapas de gestación, se observan alteraciones 9en la migración celular, la proliferación y la diferenciación de tejidos, lo que puede influir en las características físicas y neurológicas típicas del síndrome. El objetivo principal de este artículo es examinar los aspectos embriológicos del síndrome de Down, se centró en los mecanismos de no disyunción y su impacto en el desarrollo embrionario para proporcionar una comprensión más profunda de cómo las anomalías cromosómicas emergen y afectan al desarrollo prenatal. A través de una búsqueda bibliográfica exhaustiva con la utilización de PubMed y Google Scholar, en estudios publicados en los últimos cinco años (2019- 2024) para garantizar la inclusión de investigaciones actualizadas. Los hallazgos de los estudios seleccionados se sintetizaron para proporcionar una descripción general integral del conocimiento actual sobre el SD desde un punto de vista embriológico. Los avances en la genética y la biología molecular han permitido identificar los mecanismos subyacentes a estas alteraciones. Estos hallazgos ofrecen nuevas oportunidades para mejorar el diagnóstico prenatal y el manejo temprano de la condición, con un enfoque en la intervención temprana.
Palabras clave: síndrome de Down, embriología, cromosoma 21, desarrollo embrionario, diagnóstico prenatal
ABSTRACT
Down syndrome is a genetic condition characterized by the presence of an extra copy of chromosome 21. The embryology of Down syndrome has been the subject of intense research to understand how this chromosomal alteration affects embryonic and fetal development. During the early stages of gestation, alterations are observed in cell migration, proliferation, and tissue differentiation, which may influence the physical and neurological characteristics typical of the syndrome. The main objective of this article is to examine the embryological aspects of Down syndrome, focusing on the mechanisms of nondisjunction and their impact on embryonic development to provide a deeper understanding of how chromosomal abnormalities emerge and affect prenatal development. Through an exhaustive literature search using PubMed and Google Scholar, studies published in the last five years (2019-2024) were included to ensure the inclusion of up-to-date research. The findings of the selected studies were synthesized to provide a comprehensive overview of current knowledge on DS from an embryological perspective. Advances in genetics and molecular biology have made it possible to identify the mechanisms underlying these disorders. These findings offer new opportunities to improve prenatal diagnosis and early management of the condition, with a focus on early intervention.
Keywords: Down syndrome, embryology, chromosome 21, embryonic development, prenatal diagnosis
Recibido: 23/05/2025.
Aprobado: 10/09/2025.
Editor: Yasnay Jorge Saínz.
Aprobado por: Silvio Emilio Niño Escofet.
Introducción
El síndrome de Down (SD) es el trastorno genético más frecuente asociado a la discapacidad intelectual y es el resultado de la trisomía del cromosoma humano 21 (HSA21). (1) El fenotipo del SD incluye manifestaciones que afectan a varios sistemas corporales, en particular los sistemas musculoesquelético, neurológico y cardiovascular. Las personas con SD suelen presentar baja estatura, hipotonía muscular, inestabilidad atlantoaxial, densidad neuronal reducida, hipoplasia cerebelosa, discapacidad intelectual y defectos cardiacos congénitos, en particular defectos del tabique auriculoventricular. Además, las personas con SD corren un mayor riesgo de padecer ciertas enfermedades, como hipotiroidismo, enfermedades autoinmunes, apnea obstructiva del sueño, epilepsia, problemas auditivos y visuales, trastornos hematológicos (incluida la leucemia), infecciones recurrentes y enfermedad de Alzheimer (EA) de inicio precoz. (2)
Descrito por primera vez por John Langdon Down en 1866, la incidencia del síndrome de Down oscila entre 1 de cada 319 y 1 de cada 1.000 nacidos vivos y aumenta con la edad materna avanzada, con más de 200,000 casos anuales en todo el mundo. Aunque otras trisomías autosómicas son más frecuentes, suelen dar lugar a peores tasas de supervivencia posnatal, lo que convierte al síndrome de Down en la aneuploidía (trisomía 21) más frecuente entre los nacidos vivos. (3) Esto es de importancia debido a que, en 2013, se estimó que solo en EE.UU se llevaron a cabo 3400 interrupciones electivas relacionadas con el SD, lo que supuso una reducción del 33 % en el número de bebés con SD que habrían nacido ese año. (1,3) Desde una perspectiva embriológica, el síndrome de Down surge durante la meiosis, el proceso de división celular que produce los gametos (óvulos y espermatozoides). Normalmente, los cromosomas se dividen y distribuyen equitativamente en las células hijas durante la meiosis. Sin embargo, en el caso del síndrome de Down, ocurre una anomalía conocida como no disyunción, donde los cromosomas no se separan correctamente y da como resultado en una célula con un número anormal de cromosomas, lo que da lugar a un embrión con tres copias del cromosoma 21 en lugar de las dos habituales. (4)
El objetivo principal de este artículo es examinar los aspectos embriológicos del síndrome de Down, se centró en los mecanismos de no disyunción y su impacto en el desarrollo embrionario. A través de una revisión exhaustiva de la literatura y la presentación de datos actuales, este estudio busca proporcionar una comprensión más profunda de cómo las anomalías cromosómicas emergen y afectan al desarrollo prenatal. (4)
Método
Este artículo de revisión profundiza en las dimensiones embriológicas del síndrome de Down (SD), con un enfoque específico en los mecanismos de no disyunción y sus implicaciones para el desarrollo embrionario. Se realizó una búsqueda bibliográfica exhaustiva utilizando PubMed y Google Scholar, se centró en estudios publicados en los últimos cinco años (2019- 2024) para garantizar la inclusión de investigaciones actualizadas. La búsqueda utilizó palabras clave como: síndrome de Down, desarrollo embrionario, no disyunción y cromosoma 21 para identificar artículos pertinentes, con la inclusión de los operadores AND/OR en PubMed. Los criterios de selección incluyeron la relevancia para los mecanismos embriológicos en el síndrome de Down, la publicación debe ser realizada en español o inglés y que contribuya a nuevos hallazgos; mientras que se excluyeron los estudios que no abordaban directamente estos aspectos o que presentaban una nota de haber sido retraídos por la revista o el autor.(4)
La revisión extrajo datos para dilucidar los efectos de la no disyunción durante la meiosis en el desarrollo embrionario y la influencia de la trisomía 21 en el desarrollo cerebral, cardiaco y otros procesos críticos. Los hallazgos de los estudios seleccionados se sintetizaron para proporcionar una descripción general integral del conocimiento actual sobre el SD desde un punto de vista embriológico. Este proceso incluyó una evaluación crítica de la calidad del estudio y del rigor metodológico para garantizar la fiabilidad y la pertinencia de la información presentada. El objetivo era destacar los avances recientes, identificar temas recurrentes y abordar las discrepancias, para mejorar así la comprensión de los aspectos del desarrollo del síndrome de Down. (3,4)
Resultados
El cromosoma 21 humano, técnicamente conocido como Hsa21, contiene aproximadamente 225 genes codificantes de proteínas y entre 165 y 404 ARN no codificantes, que regulan la expresión génica. La sobreproducción de proteínas debida a la triplicación de genes da lugar a una serie de anomalías que afectan a múltiples sistemas corporales. (5) Esto incluye anomalías estructurales y funcionales en el corazón, el sistema nervioso y el tracto gastrointestinal. En particular, las personas con síndrome de Down presentan volúmenes reducidos en regiones cerebrales críticas como las cortezas frontal y temporal, el hipocampo, el cerebelo y el tronco encefálico. Estos cambios contribuyen a las deficiencias cognitivas y motoras que suelen asociarse a esta enfermedad. Además, existen alteraciones significativas en la conectividad neuronal, que pueden afectar a diversos aspectos de la función y el desarrollo cerebrales. (6) De esta manera se puede decir que el síndrome de Down (SD) puede presentarse de varias formas a nivel genotipo:
1. Trisomía 21 estándar (T21): Es la forma más frecuente y se da en aproximadamente el 95 % de los casos. Se caracteriza por un cariotipo de 47 cromosomas, incluido un cromosoma 21 adicional (47,XX,+21 o 47,XY,+21).
2. Trisomía 21 en mosaico: Presente en entre el 1 y el 2 % de los casos, esta forma implica una mezcla de células normales y células con un cromosoma 21 extra, esta da lugar a un cariotipo como 46,XX/47,XX,+21.
3. Translocaciones robertsonianas: Se producen en aproximadamente el 3 % de los casos e implican un tipo de reordenación cromosómica en la que los brazos largos, (segmentos q) de los cromosomas 13, 14, 15, 21 o 22 se fusionan en sus centrómeros para formar un único cromosoma grande con un solo centrómero. Para el cromosoma 21, las translocaciones robertsonianas más comunes son:
a. 46,XX,t(21;21) o 46,XY,t(21;21) (translocación entre dos cromosomas 21).
b. 46,XX,t(14;21) o 46,XY,t(14;21) (translocación entre los cromosomas 14 y 21).
c. 46,XX,t(15;21) o 46,XY,t(15;21) (translocación entre el cromosoma 15 y el 21).
4. Trisomía 21 parcial: Se trata de una forma poco frecuente, que se da en menos del 1 % de los casos, en la que solo un segmento del cromosoma 21 está triplicado.
Este síndrome va a estar asociado al brazo largo del cromosoma 21, concretamente a la banda 21q22. Dentro de esta región, la Región Crítica del Síndrome de Down (DSCR), situada entre las bandas 21q21,1 y 21q22,2, contiene aproximadamente 50 genes codificantes de proteínas junto con numerosos elementos no codificantes. Se cree que esta región engloba muchos de los factores clave responsables del fenotipo del SD. (7) Además, al evaluar la sensibilidad de los genes codificadores de proteínas del cromosoma 21 humano (HSA21) a la haploinsuficiencia (es decir, la intolerancia a los alelos heterocigotos de pérdida de función), varios genes de alta prioridad, como DYRK1A, se han relacionado con características clínicas específicas del síndrome de Down (SD).
Las investigaciones han demostrado que la sobreexpresión de DYRK1A, localizado en 21q22, desempeña un papel importante en la manifestación de los fenotipos del SD. Por ejemplo, los ratones con sobreexpresión de Dyrk1A (mBACTgDyrk1A, o Dyrk1A+/++) presentan una serie de deficiencias, como problemas de comportamiento, déficits cognitivos, anomalías motoras, cambios neuronales, defectos retinianos, problemas vasculares, anomalías gastrointestinales, deficiencias en la respuesta inmunitaria, desarrollo anómalo del timo y alteración de la función de los linfocitos T, condiciones que reflejan las observadas en la trisomía 21 humana. (8)
A esto se añade que, en la parte embriológica, la mayoría de los casos de trisomía 21 estándar son el resultado de la no disyunción cromosómica durante la meiosis materna I (Figura 1). La no disyunción paterna, que se produce durante la meiosis II, es menos frecuente pero también puede contribuir. Las translocaciones robertsonianas que afectan al cromosoma 21 pueden surgir de nuevo durante la fecundación o heredarse de uno de los progenitores. Cuando un progenitor es portador de una translocación robertsoniana, el riesgo de tener un hijo con SD es de aproximadamente el 10-15 % si la madre es la portadora, y de alrededor del 2-3 % si el padre es el portador, a pesar de que la translocación esté equilibrada y no muestre ningún exceso o déficit de material genético. (7,9)
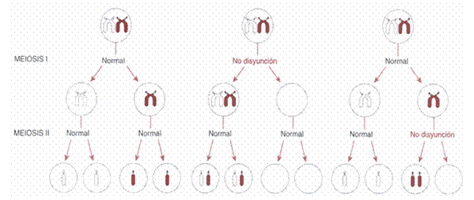
Figura 1. Las diferentes consecuencias de la no disyunción en la meiosis I y en la meiosis II. Imagen tomada del portal Fundacion Iberoamerica Down21. URL: https://www.down21.org/informacion-basica/76-que-es-el-sindrome-de-down/120trisomia-simple-por-no-disyuncion.html
La no disyunción cromosómica es el fallo en la separación de los cromosomas, que produce células hijas con un número anormal de cromosomas. La no disyunción puede producirse durante el anafase de la mitosis, la meiosis I o la meiosis II y da lugar a una distribución desigual de los cromosomas. (10) En la no disyunción mitótica, la no separación de las cromátidas hermanas debido a la inactivación de proteínas da lugar a dos células hijas aneuploides: una con 47 cromosomas y otra con 45. En la meiosis I, la no disyunción se produce en el anafase de la mitosis, en la meiosis I y en la meiosis II. En la meiosis I, la no disyunción se produce cuando las tétradas no logran separarse y producen dos células hijas haploides con un cromosoma extra (n+1) o un cromosoma ausente (n-1), que dan lugar a cuatro células hijas después de la meiosis II: dos con n+1 y dos con n-1. En la meiosis II, si las cromátidas hermanas no se separan, el resultado son cuatro células hijas, dos con un número normal de cromosomas y dos aneuploides, una con n+1 y otra con n-1. (10)
Dentro de los factores de riesgo de no disyunción se encuentra como principal la edad materna avanzada, un factor significativo en el desarrollo del síndrome de Down. Estudios han demostrado que la probabilidad de tener un hijo con síndrome de Down aumenta considerablemente en mujeres mayores de 35 años. (11) Esto se debe a que los óvulos de mujeres mayores pueden ser más propensos a errores durante la meiosis, lo cual resulta en un mayor riesgo de trisomía en la descendencia. Por otro lado, se tienen a los factores ambientales que también desempeñan un papel en el riesgo de no disyunción, aunque precisar sus efectos específicos es un reto debido a las dificultades para definir la exposición, la dosis y el momento.
Para comprender mejor la trisomía 21, es esencial categorizar el tipo de error (ya sea de origen parental o el tipo de error meiótico o mitótico) que conduce a la afección, dada su etiología heterogénea. Los factores de riesgo ambientales conocidos para la trisomía 21 incluyen el consumo de tabaco, la suplementación con ácido fólico y el uso de anticonceptivos orales, entre otros. Los estudios que abordan estos factores de riesgo y sus limitaciones, así como los posibles biomarcadores de exposición, como la longitud de los telómeros, se han revisado en otros lugares. (11) Por ejemplo, el estatus socioeconómico materno (SES) se ha relacionado específicamente con errores en la meiosis II materna. El NSE sirve como indicador indirecto de determinadas exposiciones, lo que ha llevado a un estudio de seguimiento a investigar la ocupación materna como factor de riesgo. (1,11)
En lo que corresponde al fenotipo de personas con síndrome de Down se encuentran las que presentan un conjunto de características físicas y clínicas distintivas. Las características fenotípicas incluyen: cuello corto, orejas pequeñas, puente nasal plano, pliegues epicánticos, manchas en el campo de cepillado, pliegue palmar único, boca pequeña con lengua grande y saliente, soplo cardiaco, hipotonía, escaso esfuerzo respiratorio, mala succión, distensión abdominal y vómitos (Figura #2). (12) En la mitad de los bebés con SD se observan defectos cardiacos congénitos que afectan al tabique ventricular, la tetralogía de Fallot y el canal auriculoventricular.
También existen anomalías gastrointestinales congénitas en el 12 % de los bebés con SD, como la enfermedad por reflujo gastroesofágico, la enfermedad celíaca, la fístula traqueoesofágica, el divertículo de Meckel, la atresia esofágica o duodenal, el ano imperforado, la estenosis pilórica y la enfermedad de Hirschsprung. Además de estas características físicas, los individuos con síndrome de Down suelen presentar discapacidades cognitivas y problemas de desarrollo que varían en severidad. Los desafíos en el lenguaje, el aprendizaje y la socialización son comunes, aunque el grado de afectación varía ampliamente entre los individuos. (12)

Figura 2. Dibujo de los rasgos faciales del síndrome de Down Contribución de los Centros para el Control y la Prevención de Enfermedades, Centro Nacional de Defectos Congénitos y Discapacidades del Desarrollo.
Las investigaciones indican que las personas con síndrome de Down presentan volúmenes cerebrales totales más pequeños y un patrón giral más suave y simplificado. Los estudios también han documentado una reducción de la superficie cortical y un aumento del grosor cortical en niños y adultos jóvenes con síndrome de Down. En la edad adulta media, los signos de envejecimiento prematuro del cerebro se hacen evidentes, incluida una reducción significativa del volumen en regiones clave como el córtex prefrontal, el hipocampo y el cerebelo, que son fundamentales para el habla, el aprendizaje y la memoria. (13)
Los estudios pos mortem, aunque limitados en número, han revelado la amplia variabilidad y gama de características neuropatológicas asociadas al síndrome de Down a lo largo de la vida. Estos estudios indican que las reducciones de tamaño y peso del cerebro comienzan en los períodos fetal y neonatal. Se han observado reducciones en el número de células progenitoras neuronales (CPN) proliferantes en el giro dentado, la matriz germinal del ventrículo lateral y el tercer ventrículo en tejido derivado de fetos diagnosticados con síndrome de Down (17-21 semanas de gestación) antes de las observaciones iniciales de cambios volumétricos. (14) Esta proliferación reducida parece dar lugar a un grupo más pequeño de CPN, ya que se han informado menos células SOX2+ en la zona subventricular externa (ZSV) en la corteza cerebral en desarrollo en fetos con síndrome de Down durante la mitad de la gestación (18-24 semanas), lo que potencialmente limita la producción de neuronas y glía. Por último, durante la gestación tardía, cuando se produce la expansión neocortical típica, los cerebros con síndrome de Down muestran patrones de laminaciones corticales retrasadas y desorganizadas. (14)
Los defectos del tabique auriculoventricular (AVSD, por sus siglas en inglés) abarcan una variedad de malformaciones cardíacas que se clasifican en tres subtipos principales: AVSD incompleto, AVSD transicional y AVSD completo. Los AVSD incompletos se caracterizan por anillos mitral y tricúspide separados u orificios valvulares izquierdo y derecho distintos. El desarrollo de AVSD está relacionado con la formación anormal de los cojines endocárdicos, que son cruciales para la formación del tabique. (15)
Este proceso comienza al final de la cuarta semana de desarrollo fetal cuando los cojines endocárdicos auriculoventriculares aparecen en los bordes superior e inferior del canal auriculoventricular, junto con los cojines auriculoventriculares laterales en los bordes derecho e izquierdo del canal. Una falla en la fusión de los cojines superior e inferior conduce a un canal auriculoventricular persistente, lo que resulta en un AVSD. Si bien la trisomía 21 aumenta el riesgo de defectos cardíacos congénitos (CHD, por sus siglas en inglés), no es directamente responsable de causar estos defectos. Variaciones genéticas adicionales o factores ambientales pueden contribuir aún más al riesgo de CHD.
Se han vinculado genes específicos no pertenecientes al cromosoma 21 con la susceptibilidad a diversas cardiopatías congénitas y trastornos auriculoventriculares, incluidas mutaciones en el gen CRELD1 asociado con el síndrome de Down (SD) y el trastorno auriculoventricular, así como mutaciones del gen GATA4 encontradas en familias con malformaciones cardíacas que involucran trastorno auriculoventricular. (15)
Estudios recientes sobre individuos con SD y trastorno auriculoventricular completo han identificado variantes potencialmente dañinas en varios genes, incluidos COL6A1, COL6A2, CRELD1, FBLN, FRZB y GATA5, que están involucrados en la vía VGFA. (16)
El síndrome de Down (SD) es la causa genética más común de discapacidad intelectual, resultado de la triplicación de una región genómica en el cromosoma humano 21. Otras causas pueden ser la translocación robertsoniana y el cromosoma isocromosómico o en anillo. El término isocromosoma se utiliza para describir una afección en la que dos brazos largos del cromosoma se separan juntos en lugar de que el brazo largo y el corto se separen juntos durante el desarrollo del óvulo-espermatozoide. (17)
Alrededor del 50 % de los bebés con SD nacen con un defecto cardíaco congénito (DCC), donde los defectos de los cojinetes endocárdicos son los más frecuentes, como los defectos del tabique auriculoventricular (DAV 45 %) y los defectos del tabique ventricular (DAV 35%). La presencia de DCC está relacionada con peores resultados, similar a lo que se observa en niños con DCC de desarrollo típico, que presentan déficits en las habilidades neurocognitivas, motoras y psicosociales. La gravedad y la variedad de los desafíos en el síndrome de Down (SD) se deben en parte a la sobreexpresión de ciertos genes en el cromosoma adicional y la posterior desregulación de sus vías asociadas. (18)
El cerebro fetal con síndrome de Down está reducido en peso, volumen y tamaño lineal. Se han detectado defectos de tamaño tan temprano como a los 14,7 años de edad gestacional, que afectan a las estructuras del prosencéfalo y al cerebelo y se encuentran en un rango entre -10 y -30 % en comparación con los cerebros de control. Estudios recientes de resonancia magnética en fetos vivos han permitido cuantificar las dimensiones del cerebro fetal con síndrome de Down en diferentes puntos temporales. En los resultados obtenidos por Tarui et al., (19) mencionan que los fetos con síndrome de Down (21-35 años de edad gestacional) tienen un volumen reducido de los hemisferios cerebelosos, todo el cerebelo, la placa cortical y el volumen del parénquima subcortical en comparación con los controles y la diferencia aumenta con la gestación.
Asimismo, Patkee et al. (20) encontraron una reducción en el volumen cerebral y cerebeloso en el segundo y tercer trimestre. Estos enfoques múltiples proporcionan una prueba inequívoca de que la hipotrofia cerebral es un fenotipo típico del síndrome de Down que comienza en etapas tempranas de la vida fetal, se mantiene en etapas fetales posteriores y después del nacimiento.
Conclusiones
El síndrome de Down (SD), causado por la triplicación del cromosoma 21, se manifiesta en una variedad de formas genotípicas que influyen significativamente en el fenotipo clínico y embriológico de los individuos afectados. La sobreexpresión de genes en el cromosoma 21, particularmente en la Región Crítica del Síndrome de Down (DSCR), desempeña un papel crucial en el desarrollo de anomalías estructurales y funcionales, como las observadas en el corazón y el cerebro. La no disyunción cromosómica, un fallo en la separación de los cromosomas durante la meiosis, es el mecanismo principal que da lugar a la trisomía 21, con la edad materna avanzada y factores ambientales, los cuales influyen en su aparición. Las investigaciones han revelado que las anomalías en el desarrollo cerebral comienzan en etapas tempranas de la vida fetal, ya que se manifiestan en volúmenes reducidos de regiones cerebrales críticas y alteraciones en la conectividad neuronal, lo que contribuye a las deficiencias cognitivas y motoras observadas en el SD.
Referencias bibliográficas
1. Antonarakis SE, Skotko BG, Rafii MS, Strydom A, Pape SE, Bianchi DW, et al. Down syndrome. Nat Rev Dis Primers [Internet]. 2020 [citado 11/09/2024];6(1):9. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8428796/
2. Koul AM, Ahmad F, Bhat A, Aein Q ul, Ahmad A, Reshi AA, et al. Unraveling Down Syndrome: From Genetic Anomaly to Artificial Intelligence-Enhanced Diagnosis. Biomedicines [Internet]. 2023 [citado 11/09/2024];11(12):3284. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10741860/
3. Bianchi DW, Simpson JL, McGillivray B. Genetic Disorders and the Child: A Guide to Diagnosis and Management. 6ta ed Luxemburgo:Springer; 2014.
4. Akhtar F, Bokhari SRA. Down Syndrome. En: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 [citado 12/09/2024]. Disponible en:
https://www.ncbi.nlm.nih.gov/books/NBK526016/
5. Baburamani AA, Patkee PA, Arichi T, Rutherford MA. New approaches to studying early brain development in Down syndrome. Dev Med Child Neurol. 2019 [citado 12/09/2024];61(8):867-79.
6. Windsperger K, Hoehl S. Development of Down Syndrome Research Over the Last Decades–What Healthcare and Education Professionals Need to Know. Front Psychiatry. 2021 [citado 12/09/2024];12:749046. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8712441/
7. Rondal JA. From the lab to the people: major challenges in the biological treatment of Down syndrome. AIMS Neurosci . 2021 [citado 12 /09/ 2024]; 8(2):284-94. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7940110/
8. Ishihara K. Genes Associated with Disturbed Cerebral Neurogenesis in the Embryonic Brain of Mouse Models of Down Syndrome. Genes (Basel). 2021 [citado 12 /09/ 2024];12(10):1598. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8535956/
9. PLAIASU V. Down Syndrome – Genetics and Cardiogenetics. Maedica (Bucur). 2017 [citado 12/09/2024];12(3):208-13. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5706761/
10. Gottlieb SF, Tupper C, Kerndt CC, Tegay DH. Genetics, Nondisjunction. En: StatPearls Treasure Island (FL): StatPearls Publishing; 2024 [citado 12/09/2024]. Disponible en: http://www.ncbi.nlm.nih.gov/books/NBK482240/
11. Blanco-Montaño A, Ramos-Arenas M, Yerena-Echevarría BA, Miranda-Santizo LD, Ríos-Celis AL, Dorantes-Gómez AT, et al. Factores de riesgo en el origen del síndrome de Down. Rev Med Inst Mex Seguro Soc. 2023 [citado 12/09/2024];61(5):638-44. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10599770/
12. Rafii MS, Kleschevnikov AM, Sawa M, Mobley WC. Down syndrome. Handb Clin Neurol. 2019;167:321- 36.
13. Halevy T, Biancotti JC, Yanuka O, Golan-Lev T, Benvenisty N. Molecular Characterization of Down Syndrome Embryonic Stem Cells Reveals a Role for RUNX1 in Neural Differentiation. Stem Cell Reports. 2016 [citado 12/09/2024];7(4):777-86. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5063584/
14. Klein JA, Haydar TF. Neurodevelopment in Down syndrome: Concordance in humans and models. Front Cell Neurosci. 2022 [citado 12/09/2024];16:941855. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9334873/
15. Dimopoulos K, Constantine A, Clift P, Condliffe R, Moledina S, Jansen K, et al. Cardiovascular Complications of Down Syndrome: Scoping Review and Expert Consensus. Circulation. 2023 [citado 12/09/2024];147(5):425-41. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9977420/
16. Sanaa B, Abdenasser D, Ayoub EH. Congenital heart disease and Down syndrome: various aspects of a confirmed association. Cardiovasc J Afr. 2016 [citado12/09/2024];27(5):287-90. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5370349/
17. Asim A, Kumar A, Muthuswamy S, Jain S, Agarwal S. “Down syndrome: an insight of the disease”. J Biomed Sci. 2015 [citado 12/09/2024];22(1):41. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4464633/
18. CILHOROZ BT, RECENO CN, HEFFERNAN KS, DERUISSEAU LR. Cardiovascular Physiology and Pathophysiology in Down Syndrome. Physiol Res. 2022 [citado12/09/2024];71(1):1-16. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8997681/
19. Tarui T, Im K, Madan N, Madankumar R, Skotko BG, Schwartz A, et al. Quantitative MRI Analyses of Regional Brain Growth in Living Fetuses with Down Syndrome. Cereb Cortex. 2020 [citado 12/09/2024];10;30(1):382-390. Disponible en: https://pmc.ncbi.nlm.nih.gov/articles/PMC7029684/pdf/bhz094.pdf
20. Patkee PA, Baburamani AA, Kyriakopoulou V, Davidson A, Avini E, Dimitrova R, et al. Early alterations in cortical and cerebellar regional brain growth in Down Syndrome: An in vivo fetal and neonatal MRI assessment. Neuroimage Clin. 2019 [citado 12 /09/2024];25:102139. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6938981/
Declaración de conflicto de intereses
Los autores no declaran conflicto de intereses
Contribución de autoría
Los autores participaron en igual medida en la curación de datos, análisis formal, investigación, metodología, administración del proyecto, recursos, software, supervisión, validación, visualización, redacción – borrador original y redacción – revisión y edición.
![]() Los
artículos de la Revista Correo Científico Médico
perteneciente a la Universidad de Ciencias Médicas de Holguín se comparten bajo
los términos de la Licencia Creative Commons Atribución 4.0 Internacional
Email: publicaciones@infomed.sld.cu
Los
artículos de la Revista Correo Científico Médico
perteneciente a la Universidad de Ciencias Médicas de Holguín se comparten bajo
los términos de la Licencia Creative Commons Atribución 4.0 Internacional
Email: publicaciones@infomed.sld.cu